PMDA(医薬品医療機器総合機構)の不思議-特集1「医薬品審査」
では、実際の審査はどのように行われているのだろうか。
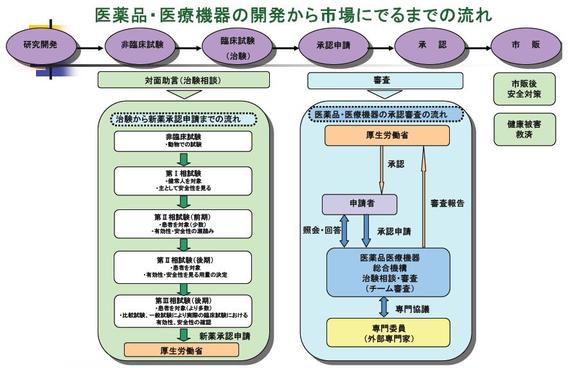
製薬企業が新しく開発した医薬品が、私たちの手に渡るようになるには、まず製薬企業が開発した医薬品について動物実験や人体に対する臨床試験(治験)を行う必要がある。企業側は「実験の結果、このように安全性や有効性が確認されたので、患者さんにちゃんと効く薬です。承認して下さい」(実際に承認するのは国)として、その申請書類や試験結果に関するデータが記載された資料をPMDAに提出する。これが「一つの医薬品でダンボール箱3箱分ぐらい」(PMDA審査員)もある分量で、紙かデータの形で提出される。審査員は、この膨大な書類に目を通し、試験結果などを見ながら「安全」や「有効」を導き出しているプロセスや論理展開に矛盾がないか、根拠がおかしくないかといったことなどについて検証する。
現在の審査部門では346人の職員が働いている。「抗悪性腫瘍剤分野」「血液製剤分野」「生物製剤分野」など、薬の種類によって審査員がチーム分けされており、それぞれに薬学、医学、獣医学、生物統計学の専門家として、医師や薬剤師などの資格を持った審査員が10人ずつ程度配置されている。2007年度には81件の医薬品が承認され、一つの医薬品を審査するのに約1年8か月かかっていた。
また、審査される医薬品の担当審査員とは別の職員になるが、医薬品の有効性や安全性を示す実験について、国が定めた基準の下にきちんと実施されているかどうかを、国内外に出張して現地調査することもある。さらに、申請された医薬品を製造できる体制が企業内に整っているかも実地で調査する。
PMDAでの審査を受けた後、厚労省の「薬事・食品衛生審議会」に審査結果が提出され、承認を受ければ、医薬品は晴れてこの世に出ていけるお墨付きをもらったことになる。
(資料:PMDA提供)
このように、PMDAは企業と国の中間に位置し、企業側の言うなりに医薬品・医療機器が社会に出てしまわないよう、一定の「規制」をかけるという役割を担っている。その規制が「審査」ということになり、企業側が医薬品を売りたいがために間違ったことを言っていないか、また意図せずともおかしなプロセスを経ていないかなどについて、審査員が自身の知識やこれまで培ってきた審査経験などに基づいて確かめることで、申請された医薬品が本当に安全で有効なのかを検証していくことになる。つまり、審査は個々の審査員の経験や勘に頼るアナログな人力作業なのだ。
このため、審査員は日々世界中から上がってくる医薬研究や医療技術開発の論文発表などを確認したり、国際的な学会に出席したりするなどして、日進月歩の知識を蓄積していかなければ、新しく出されるデータを把握していけなくなってしまう。また、海外で医薬品について何か問題があった時に、その国のPMDAに当たる機関(米国ならFDA(米食品医薬品局)、EUはEMEA(欧州医薬品庁))がどう対応したかも参考にするので、諸外国の動向も見逃せない。PMDAで麻酔用薬などを審査するチームのリーダーを務める宇山佳明氏(薬学博士)は「審査される医薬品の情報についての交通整理が審査員の仕事。常に新しい情報をアップデートすることが必要になる」と話す。
また、一人の審査員が複数の医薬品を担当しているため、審査の質や信頼性の担保が課題になる。宇山氏は、「医薬品の種類によってチームに分かれているので、チーム内で情報共有し、気付いたことがあれば指摘し、話し合う」と話す。多くの経験を持つスタッフが関わることで、少人数の判断に依存しないように工夫しているということだ。
ただ、企業側の主張や、臨床試験などがルールにのっとったものかについての確認には多くのガイドラインがあるためそれを参考にできるが、その医薬品・医療機器が、今の日本の社会の中でどれほど必要とされ、それがあることで社会にどういう影響を与えるかといったことなど、「そもそも承認審査に値する医薬品・医療機器なのかどうか」ということを評価する基準はないのが現状だ。
そんなPMDAに、大きな課題がのしかかっている。
